Evolution of International/Global Clone 1
Authors: María Paula Quiroga1, 2, Verónica E. Álvarez1, and Daniela Centron1
1Laboratorio de Investigaciones en Mecanismos de Resistencia a Antibióticos, Instituto de Investigaciones en Microbiología y Parasitología Médica, Facultad de Medicina, Universidad de Buenos Aires - Consejo Nacional de Investigaciones Científicas y Tecnológicas (IMPaM, UBA-CONICET), Argentina (valvarez@gmail.com; dcentron@gmail.com)
2Nodo Bioinformático del Instituto de Investigaciones en Microbiología y Parasitología Médica, Facultad de Medicina, Universidad de Buenos Aires - Consejo Nacional de Investigaciones Científicas y Tecnológicas (IMPaM, UBA-CONICET), Argentina (quirogamp@gmail.com)
Reviewer: Raffaele Zarrilli
Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II, Naples, Italy (rafzarri@unina.it)
Molecular typing of Acinetobacter baumannii International/Global clone 1
Acinetobacter baumannii Global Clone 1 (GC1), also known as International Clone I, has since the 1970s been emerging as an important superbug able to rapidly develop resistance to a wide range of antibiotics including carbapenems (Holt et al., 2016), being one of the prevalent producers of OXA-23 in many countries (Cardoso et al., 2016; Hamed et al., 2022). GC1 is currently correlated with 69 sequence types (Table 1a, as of December 2022), forming a monophyletic clonal complex (CC), designated CC1P, according to the Pasteur Multi Locus Sequence typing (MLST) scheme and using one locus variant as the maximum number of differences between nodes (Diancourt et al., 2010; Feil et al., 2004; Gaiarsa et al., 2019; Karah et al., 2021). The Oxford MLST scheme (Bartual et al., 2005), known to have a higher discriminatory power, has so far assigned 111 sequence types to GC1 (Table 1b), which are grouped into a non-monophyletic CC, previously called CC109B and recently re-designed to CC231Ox (Hamidian et al., 2017), with at least two clearly separated clades (Gaiarsa et al., 2019). Additional phylogenetic analysis of GC1 revealed two major clades, C1 and C2 (Holt et al., 2016; Hamidian and Hall, 2021), which include five lineages (L1–L5), and four separate single-isolate lineages outside C1 and C2 (Koong et al., 2021), corresponding two of them to environmental samples. Each lineage showed specific features: L1 included geographically and genetically diverse genomes associated with the AbaR0-type resistance islands, L2 was associated with strains isolated or linked to Middle East region with AbaR4 resistance islands, L3 grouped strains isolated from India, Afghanistan and Mexico containing either AbaR4 or its variants as well as Tn6022::ISAba42, L4 assembled strains isolated from Eastern Europe, USA, Egypt and Morocco associated with Tn6022 or its variants, and L5 accumulated strains mostly isolated from USA, also associated with Tn6022 or its variants (Koong et al., 2021).
When analyzing the contribution of the accessory genome to the evolution of GC1, some genetic traits were found located at the same loci as “sedentary” modules such as both the case of a CRISPR-Cas system acquired before clonal diversification, and the AbaR0-type genomic island with signs of microevolution in C1. The prophage YMC/09/02/B1251_ABA_BP was found to be “mobile” since, although it was shared by all five lineages, it showed high intrinsic microevolution as well as mobility to different insertion sites. Interestingly, a wide variety of Insertion Sequences (IS), probably acquired by the flow of plasmids related to Rep_3 superfamily was found. These IS showed dissimilar genomic location amongst GC1 genomes presumably associated with promptly niche adaptation. On the other hand, a type VI secretion system and three efflux pumps were subjected to deep processes of genomic loss in A. baumannii but not in GC1 (Álvarez et al., 2020).
Global epidemiology of GC1
More extensive phylogenetic studies, including 298 GC1 genomes selected from the total of 512 genomes deposited in GenBank (until June 2021) from different sources covering a period of 39 years (1982-2021) showed that L1 is widespread in the 6 continents found in human -colonizing and infecting samples, in abiotic surfaces of hospitals, in animals and in environmental habitats (Figure 1). Although this dissemination may be due to a greater representation of samples, also evidences that GC1 is not restricted to cause serious human infections, but also environmental GC1 strains interact with human activities that may be implicated in the disease burden of this successful pandemic clone.
Sub-lineages with strains geographically related (Koong et al., 2021), were even isolated with a time difference of more than two decades (Figure 1). As an example of this microevolution along time are the cases of Argentina (L1, 1994-2021), Australia (L1, 1995-2011), Brazil (L1, 2008-2020), Switzerland (L1, 2006-2015), Germany (L1, 2006-2015), Singapore (L1, 2004-2011), Egypt (L1, 2010-2015), and USA (L2, 2003-2011 and L4, 2003-2018). This shows that these sub-lineages from different hospitals are able to adapt, to effectively establish and to persist along time, evidencing in small scale how successfully global clones evolve under extreme antimicrobial pressure.
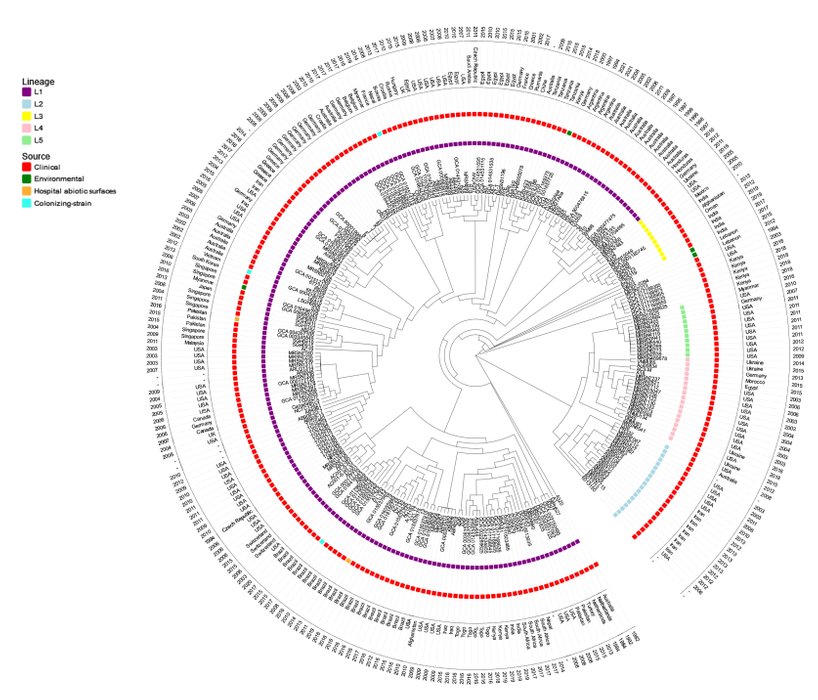
Figure 1. Phylogenetic tree of A. baumannii GC1 strains. A total of 298 A. baumannii GC1 genomes were selected from the 512 genomes deposited in GenBank until June 2021, which included clinical strains from the 6 continents isolated along time, and all the environmental strains identified. A maximum likelihood phylogenetic tree was reconstructed from a core genome alignment using the genome sequences downloaded from GenBank. The sequences were mapped to the A. baumannii A1 strain, which was used as a reference (GenBank accession number CP010781.1) using the Snippy software (v.2.0) (available at https://github.com/tseemann/snippy) to generate a whole- genome alignment. High-quality variant sites were called using SAMtools (v1.3.1.24). Single nucleotide differences (SNDs) located in recombinant regions were identified and removed using the Gubbins (v2.1.025) program with default parameters. A maximum-likelihood phylogenetic tree was inferred from the resulting alignment using FastTree (v2.1) (available at http://www.microbesonline.org/fasttree/) with the Generalized Time Reversible (GTR) Gamma model of nucleotide substitution using 1000 bootstraps. The A. baumannii GC2 strain A320 (RUH134) (GenBank accession number CP032055.1), was used as the outgroup for phylogenetic analysis. Lineage and sub-lineage assignments were done according to Koong et al., 2021.
References
Holt K, Kenyon JJ, Hamidian M, Schultz MB, Pickard DJ, Dougan G, Hall R. Five decades of genome evolution in the globally distributed, extensively antibiotic-resistant Acinetobacter baumannii global clone 1. Microb Genom. 2016 Feb 23;2(2):e000052.
Erratum in: Microb Genom. 2019 Jul;5(7)
PMID: 28348844
Cardoso JP, Cayô R, Girardello R, Gales AC. Diversity of mechanisms conferring resistance to β-lactams among OXA-23-producing Acinetobacter baumannii clones. Diagn Microbiol Infect Dis. 2016 May;85(1):90-7.
doi: 10.1016/j.diagmicrobio.2016.01.018
PMID: 26971181
Hamed SM, Hussein AFA, Al-Agamy MH, Radwan HH, Zafer MM. Genetic configuration of genomic resistance islands in Acinetobacter baumannii clinical isolates From Egypt. Front Microbiol. 2022 Jul 22;13:878912.
doi: 10.3389/fmicb.2022.878912
PMID: 35935207
Diancourt L, Passet V, Nemec A, Dijkshoorn L, Brisse S. The population structure of Acinetobacter baumannii: expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS One. 2010 Apr 7;5(4):e10034.
doi: 10.1371/journal.pone.0010034
PMID: 20383326
Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004 Mar;186(5):1518-30.
doi: 10.1128/JB.186.5.1518-1530.2004
PMID: 14973027
Karah N, Wai SN, Uhlin BE. CRISPR-based subtyping to track the evolutionary history of a global clone of Acinetobacter baumannii. Infect Genet Evol. 2021 Jun;90:104774.
doi: 10.1016/j.meegid.2021.104774
PMID: 33618003
Bartual SG, Seifert H, Hippler C, Luzon MA, Wisplinghoff H, Rodríguez-Valera F. Development of a multilocus sequence typing scheme for characterization of clinical isolates of Acinetobacter baumannii. J Clin Microbiol. 2005 Sep;43(9):4382-90.
doi: 10.1128/JCM.43.9.4382-4390.2005
Erratum in: J Clin Microbiol. 2007 Jun;45(6):2101
PMID: 16145081
Hamidian M, Nigro SJ, Hall RM. Problems with the Oxford multilocus sequence typing scheme for Acinetobacter baumannii: Do sequence type 92 (ST92) and ST109 exist? J Clin Microbiol. 2017 Jul;55(7):2287-2289.
doi: 10.1128/JCM.00533-17
PMID: 28490493
Gaiarsa S, Batisti Biffignandi G, Esposito EP, Castelli M, Jolley KA, Brisse S, Sassera D, Zarrilli R. Comparative analysis of the two Acinetobacter baumannii multilocus sequence typing (MLST) schemes. Front Microbiol. 2019 May 3;10:930.
PMID: 31130931
Hamidian M, Hall RM. Dissemination of novel Tn7 family transposons carrying genes for synthesis and uptake of fimsbactin siderophores among Acinetobacter baumannii isolates. Microb Genom. 2021 Mar;7(3):mgen000548.
PMID: 33749577
Koong J, Johnson C, Rafei R, Hamze M, Myers GSA, Kenyon JJ, Lopatkin AJ, Hamidian M. Phylogenomics of two ST1 antibiotic-susceptible non-clinical Acinetobacter baumannii strains reveals multiple lineages and complex evolutionary history in global clone 1. Microb Genom. 2021 Dec;7(12):000705.
PMID: 34874246
Álvarez VE, Quiroga MP, Galán AV, Vilacoba E, Quiroga C, Ramírez MS, Centrón D. Crucial role of the accessory genome in the evolutionary trajectory of Acinetobacter baumannii global clone 1. Front Microbiol. 2020 Mar 18;11:342.
PMID: 32256462