Global Epidemiology
Author: Valeria Mateo-Estrada and Santiago Castillo-Ramírez
Programa de Genómica Evolutiva, Centro de Ciencias Genómicas, Universidad Nacional Autónoma de México, Cuernavaca, México (vmateo@lcg.unam.mx; iago@ccg.unam.mx)
Reviewer: Paul G Higgins
Institute for Medical Microbiology, Immunology and Hygiene, University of Cologne, Cologne, Germany
(paul.higgins@uni-koeln.de)
Molecular epidemiology of Acinetobacter baumannii
Acinetobacter baumannii is a frequent cause of infections and epidemic outbreaks in hospitals worldwide (Tomaschek et al., 2016). To better understand and tackle A. baumannii, global epidemiology studies are essential to identifying epidemic clones and gaining insights into the most important lineages affecting humans. The initial gold standard for studying the molecular epidemiology of this bacterium was pulsed-field gel electrophoresis (PFGE) (Seifert et al., 2005), a method that relies on comparing restriction patterns of DNA. PFGE has been particularly useful for its highly discriminatory power in identifying epidemic outbreaks and studying genomic diversity (Bartual et al., 2005; Tomaschek et al., 2016; Higgins et al., 2017). However, PFGE genotypes (pulse types) are difficult to compare and store in databases, limiting their power to local studies or single laboratories. Methods based on direct DNA sequence comparison at the gene or genome level can overcome these limitations. Genotypes generated by these methods could be stored and shared in databases allowing comparisons at the international level (Bartual et al., 2005; Gaiarsa et al., 2019).
Multilocus sequence typing (MLST) is the most widely used sequence comparative method for population analysis of bacterial pathogens (Bartual et al., 2005; Diancourt et al., 2010; Tomaschek et al., 2016; Castillo-Ramírez and Graña-Miraglia, 2019). MLST relies on partial sequencing of alleles from 7 housekeeping genes, and then a number, the sequence type (ST), is assigned to the allelic profile. Two MLST schemes have been developed for A. baumannii, the Oxford (Bartual et al., 2005) and Pasteur (Diancourt et al., 2010) schemes. The schemes share some alleles but have different levels of resolution (Tomaschek et al., 2016; Castillo-Ramírez and Graña-Miraglia, 2019; Gaiarsa et al., 2019). The Pasteur scheme has less resolution than the Oxford scheme but is more suitable for studying clonal lineages and identifying isolates related to the primary clones of A. baumannii (Diancourt et al., 2010; Castillo-Ramírez and Graña-Miraglia, 2019; Gaiarsa et al., 2019). Whereas, the Oxford scheme is considered as having more resolution (Tomaschek et al., 2016; Castillo-Ramírez and Graña-Miraglia, 2019). However, some genes of this scheme showed recombination signals or even paralogy (Tomaschek et al., 2016; Gaiarsa et al., 2019). Moreover, in some cases, this scheme does not produce monophyletic groups (isolates from the same ST do not cluster together) (Castillo-Ramírez and Graña-Miraglia, 2019).
More discriminatory genotyping methods that use whole genome sequencing are more appropriate for bacteria with highly dynamic genomes, such as A. baumannii (Castillo-Ramírez and Graña-Miraglia, 2019; Gaiarsa et al., 2019; Mateo-Estrada et al., 2021; Castillo-Ramírez, 2022a). Most genomic epidemiology studies focus on analyzing single nucleotide polymorphisms (SNPs) in the core genome (Castillo-Ramírez, 2022a) – regions of the bacterial genome present in all the isolates considered - along with clinical records (i.e. patient movement) to study transmission. Furthermore, an MLST based on the allelic variants in core genes (cgMLST) provides highly discriminatory data for examining A. baumannii outbreaks (Higgins et al., 2017). cgMLST is less affected by recombination events because it relies upon identifying the combination of allelic variants of the core genes rather than a total number of SNPs (Higgins et al., 2017). Importantly, the allelic profiles can be stored and shared via databases, just like with the traditional 7-gene MLST (Higgins et al., 2017). A novel approach recently suggested that the accessory genome – regions of the genome present in some but not all the isolates – can provide valuable information along with the core genome to study transmission dynamics of isolates, especially in micro-timescales where SNPs in the core genome have not accumulated (Mateo-Estrada et al., 2021). These methods have undoubtedly provided information of paramount importance on the global epidemiology of this bacterium.
A few International Clones
The molecular methods mentioned above have unveiled that particular A. baumannii clones are predominant in clinical settings from different parts of the world. Early studies identified three principal clones in European countries, so they initially received the name of European clones 1, 2, and 3. Further investigations identified isolates of these clones internationally, so they were designated as worldwide clonal lineages, global clones, or international clones. To date, nine international clones (ICs) named IC 1 to 9 have been described, although there is evidence for at least five more (Higgins et al., 2010; Tomaschek et al., 2016). Under an MLST framework, the isolates from an IC all come from the same clonal complex (CC). Considering the Pasteur scheme, the 9 ICs have been assigned to CCs 1, 2, 3, 15, 79, 78, 25, 10, and 85, respectively (Diancourt et al., 2010; Tomaschek et al., 2016; Higgins et al., 2017; Levy-Blitchtein et al., 2018; Gaiarsa et al., 2019). The Oxford scheme complicates IC assignation because of the extensive CCs described and because some, but not all IC’s, have two gdhB alleles that are only identified using whole genome sequences; importantly, by PCR only one allele will amplify. Some ICs, particularly IC2, are widely distributed in different continents (Higgins et al., 2010), while others like IC5 have been found predominantly in Latin America (Higgins et al., 2010; Graña-Miraglia et al., 2020; Mateo-Estrada et al., 2021).
Based on a core-genome of 2390 alleles, the international clones are separated by almost 2000 allelic differences (Bartual et al., 2005; Higgins et al., 2017). Isolates that do not cluster with the ICs also differ from each other by a similar distance. Thus, A. baumannii genomes have particular “flavours” and this is reflected in some lineage-specific alleles that include the intrinsic blaOXA-51-like, which can be used as a crude method to assign isolates to the IC’s (Turton et al., 2007; Zander et al., 2012). The ICs' origin remains unknown (Higgins et al., 2010), but investigations point out that these successful lineages followed a rapid clonal expansion, and little time may have elapsed for diversification (Diancourt et al., 2010). Generally speaking, isolates from the ICs tend to cause epidemic outbreaks and are multidrug-resistant (MDR) (Diancourt et al., 2010). However multidrug resistance is not only found in the ICs as there are multiple examples of resistant singletons (Higgins et al., 2010), but these isolates tend to cause sporadic infections and are generally more susceptible to antibiotics (Diancourt et al., 2010). Other factors are at play that allow the ICs to persist in the hospital environment but these are currently poorly understood. Furthermore, although the ICs were defined with human-related A. baumannii isolates, some strains collected from different hosts and environments are also related to these clones (Ewers et al., 2017; Püntener-Simmen et al., 2019; Castillo-Ramírez, 2022b).
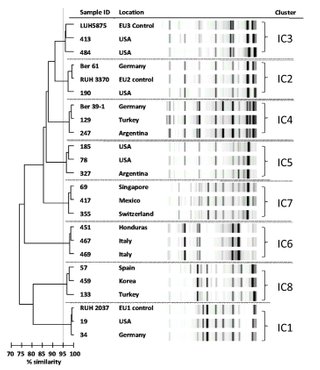
Dendrogram and computer-generated image of rep-PCR banding patterns showing representatives of the eight international clones and their country of isolation (adapted from Higgins et al., 2010).
(Figure credit: Carina Müller, mauscript in preparation).
Beyond the clinical settings
Although there is a good amount of information on human clinical isolates, there is little information on the clones potentially circulating in animals and the environment (Püntener-Simmen et al., 2019). Epidemiological studies on non-human isolates have mainly focused on companion animals like dogs and cats, where a few isolates showed a relation to ICs 1, 2, 7, and 8 (Ewers et al., 2017; Püntener-Simmen et al., 2019). Nevertheless, the majority of the studies sampled animals in veterinary clinics, which are similar to the hospital environments and could explain the similarity between these and human-related isolates; hence future studies on companion animals should also consider healthy specimens (Castillo-Ramírez, 2022b). In contrast, studies conducted in livestock (cattle and pigs) (Mateo-Estrada et al., 2022) and wildlife (white storks) (Wilharm et al., 2017) have identified non-clonal isolates unrelated to clinical isolates. Furthermore, only a few investigations have collected A. baumannii isolates from soils and plants, and these are similar to those from wildlife i.e. non-clonal (Singh et al., 2013; Repizo et al., 2017; Furlan et al., 2018). Clearly, more research on animal and environmental isolates is required, but these studies highlight that the genetic diversity of this species may be underrated. Future epidemiological studies on A. baumannii should consider animal and environmental isolates because transmission may occur between isolates from different origins (Eveillard et al., 2013). Notably, some of these non-clinical isolates may carry antibiotic-resistance genes clinically relevant for humans (Hernández-González and Castillo-Ramírez, 2020; Zheng et al., 2020; Hernández-González et al., 2022). For these reasons, A. baumannii should be analyzed under the One Health perspective (Hernández-González and Castillo-Ramírez, 2020; Castillo-Ramírez, 2022b), a multidisciplinary effort where human, animal, and environmental health are interconnected (Hernández-González and Castillo-Ramírez, 2020). Broadening the scope of the epidemiological studies could provide valuable information on the probable origin and the habitats inhabited by this bacterium.
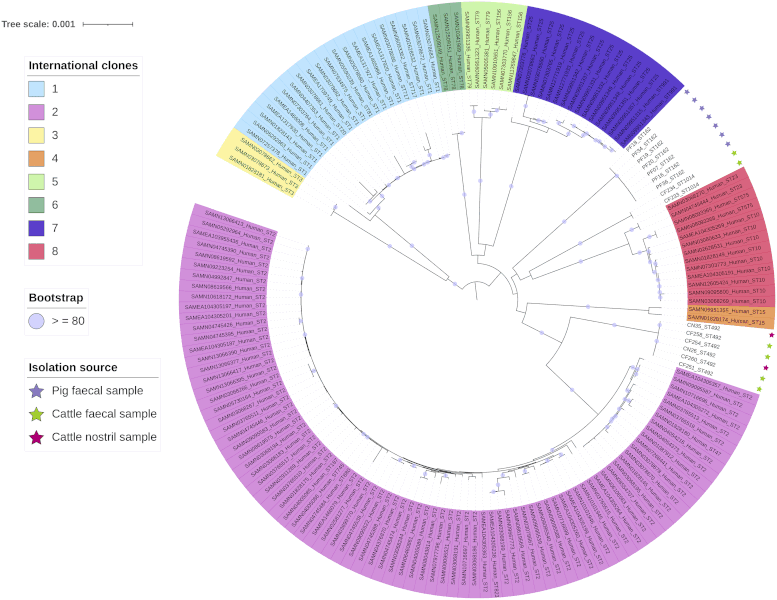
Core genome maximum likelihood phylogeny of animal isolates and 148 clinical isolates from eight international clonal lineages. The major international clones are highlighted with different colors on the labels. Pig isolates are denoted with violet stars, whereas cattle isolates are shown with green (fecal samples) and rosy (nostril samples) stars. The tree scale is the number of substitutions per site and bootstrap values higher (or equal to) 80 are depicted with violet circles at the internal nodes of the phylogeny (Mateo-Estrada et al., 2022).
References
Tomaschek F, Higgins PG, Stefanik D, Wisplinghoff H, Seifert H. Head-to-head comparison of two multi-locus sequence typing (MLST) schemes for characterization of Acinetobacter baumannii outbreak and sporadic isolates. PLoS One. 2016 Apr 12;11(4):e0153014.
doi: 10.1371/journal.pone.0153014
PMID: 27071077
Seifert H, Dolzani L, Bressan R, van der Reijden T, van Strijen B, Stefanik D, Heersma H, Dijkshoorn L. Standardization and interlaboratory reproducibility assessment of pulsed-field gel electrophoresis-generated fingerprints of Acinetobacter baumannii. J Clin Microbiol. 2005 Sep;43(9):4328-35.
doi: 10.1128/JCM.43.9.4328-4335.2005
PMID: 16145073
Bartual SG, Seifert H, Hippler C, Luzon MA, Wisplinghoff H, Rodríguez-Valera F. Development of a multilocus sequence typing scheme for characterization of clinical isolates of Acinetobacter baumannii. J Clin Microbiol. 2005 Sep;43(9):4382-90.
doi: 10.1128/JCM.43.9.4382-4390.2005
PMID: 16145081
Erratum in: J Clin Microbiol. 2007 Jun;45(6):2101
Higgins PG, Prior K, Harmsen D, Seifert H. Development and evaluation of a core genome multilocus typing scheme for whole-genome sequence-based typing of Acinetobacter baumannii. PLoS One. 2017 Jun 8;12(6):e0179228.
doi: 10.1371/journal.pone.0179228
PMID: 28594944
Gaiarsa S, Batisti Biffignandi G, Esposito EP, Castelli M, Jolley KA, Brisse S, Sassera D, Zarrilli R. Comparative analysis of the two Acinetobacter baumannii multilocus sequence typing (MLST) schemes. Front Microbiol. 2019 May 3;10:930.
PMID: 31130931
Diancourt L, Passet V, Nemec A, Dijkshoorn L, Brisse S. The population structure of Acinetobacter baumannii: expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS One. 2010 Apr 7;5(4):e10034.
doi: 10.1371/journal.pone.0010034
PMID: 20383326
Castillo-Ramírez S, Graña-Miraglia L. Inaccurate multilocus sequence typing of Acinetobacter baumannii. Emerg Infect Dis. 2019 Jan;25(1):186-187.
PMID: 30561303
Mateo-Estrada V, Fernández-Vázquez JL, Moreno-Manjón J, Hernández-González IL, Rodríguez-Noriega E, Morfín-Otero R, Alcántar-Curiel MD, Castillo-Ramírez S. Accessory genomic epidemiology of cocirculating Acinetobacter baumannii clones. mSystems. 2021 Aug 31;6(4):e0062621.
doi: 10.1128/mSystems.00626-21
PMID: 34282943
Castillo-Ramírez S. Beyond microbial core genomic epidemiology: towards pan genomic epidemiology. Lancet Microbe. 2022a Apr;3(4):e244-e245.
doi: 10.1016/S2666-5247(22)00058-1
PMID: 35544058
Higgins PG, Dammhayn C, Hackel M, Seifert H. Global spread of carbapenem-resistant Acinetobacter baumannii. J Antimicrob Chemother. 2010 Feb;65(2):233-8.
doi: 10.1093/jac/dkp428
PMID: 19996144
Erratum in: J Antimicrob Chemother. 2010 Jun;65(6):1317
Levy-Blitchtein S, Roca I, Plasencia-Rebata S, Vicente-Taboada W, Velásquez-Pomar J, Muñoz L, Moreno-Morales J, Pons MJ, Del Valle-Mendoza J, Vila J. Emergence and spread of carbapenem-resistant Acinetobacter baumannii international clones II and III in Lima, Peru. Emerg Microbes Infect. 2018 Jul 4;7(1):119.
doi: 10.1038/s41426-018-0127-9
PMID: 29970918
Graña-Miraglia L, Evans BA, López-Jácome LE, Hernández-Durán M, Colín-Castro CA, Volkow-Fernández P, Cevallos MA, Franco-Cendejas R, Castillo-Ramírez S. Origin of OXA-23 variant OXA-239 from a recently emerged lineage of Acinetobacter baumannii international clone V. mSphere. 2020 Jan 8;5(1):e00801-19.
PMID: 31915222
Turton JF, Gabriel SN, Valderrey C, Kaufmann ME, Pitt TL. Use of sequence-based typing and multiplex PCR to identify clonal lineages of outbreak strains of Acinetobacter baumannii. Clin Microbiol Infect. 2007 Aug;13(8):807-15.
doi: 10.1111/j.1469-0691.2007.01759.x
PMID: 17610600
Zander E, Nemec A, Seifert H, Higgins PG. Association between β-lactamase-encoding blaOXA-51 variants and DiversiLab rep-PCR-based typing of Acinetobacter baumannii isolates. J Clin Microbiol. 2012 Jun;50(6):1900-4.
doi: 10.1128/JCM.06462-11
PMID: 22422849
Ewers C, Klotz P, Leidner U, Stamm I, Prenger-Berninghoff E, Göttig S, Semmler T, Scheufen S. OXA-23 and ISAba1-OXA-66 class D β-lactamases in Acinetobacter baumannii isolates from companion animals. Int J Antimicrob Agents. 2017 Jan;49(1):37-44.
doi: 10.1016/j.ijantimicag.2016.09.033
PMID: 27890443
Püntener-Simmen S, Zurfluh K, Schmitt S, Stephan R, Nüesch-Inderbinen M. Phenotypic and genotypic characterization of clinical isolates belonging to the Acinetobacter calcoaceticus-Acinetobacter baumannii (ACB) complex isolated from animals treated at a veterinary hospital in Switzerland. Front Vet Sci. 2019 Feb 5;6:17.
PMID: 30805352
Castillo-Ramírez S. Zoonotic Acinetobacter baumannii: the need for genomic epidemiology in a One Health context. Lancet Microbe. 2022b Sep 20;3(12):e895–6.
doi: 10.1016/S2666-5247(22)00255-5
PMID: 36150399
Mateo-Estrada V, Vali L, Hamouda A, Evans BA, Castillo-Ramírez S. Acinetobacter baumannii sampled from cattle and pigs represent novel clones. Microbiol Spectr. 2022 Aug 31;10(4):e0128922.
doi: 10.1128/spectrum.01289-22
PMID: 35766493
Wilharm G, Skiebe E, Higgins PG, Poppel MT, Blaschke U, Leser S, Heider C, Heindorf M, Brauner P, Jäckel U, Böhland K, Cuny C, Łopińska A, Kaminski P, Kasprzak M, Bochenski M, Ciebiera O, Tobółka M, Żołnierowicz KM, Siekiera J, Seifert H, Gagné S, Salcedo SP, Kaatz M, Layer F, Bender JK, Fuchs S, Semmler T, Pfeifer Y, Jerzak L. Relatedness of wildlife and livestock avian isolates of the nosocomial pathogen Acinetobacter baumannii to lineages spread in hospitals worldwide. Environ Microbiol. 2017 Oct;19(10):4349-4364.
PMID: 28925528
Singh NK, Kumar S, Raghava GP, Mayilraj S. Draft genome sequence of Acinetobacter baumannii strain MSP4-16. Genome Announc. 2013 Apr 4;1(2):e0013713.
PMID: 23558533
Repizo GD, Viale AM, Borges V, Cameranesi MM, Taib N, Espariz M, Brochier-Armanet C, Gomes JP, Salcedo SP. The environmental Acinetobacter baumannii isolate DSM30011 reveals clues into the preantibiotic era genome diversity, virulence potential, and niche range of a predominant nosocomial pathogen. Genome Biol Evol. 2017 Sep 1;9(9):2292-2307.
doi: 10.1093/gbe/evx162
PMID: 28934377
Furlan JPR, Pitondo-Silva A, Stehling EG. New STs in multidrug-resistant Acinetobacter baumannii harbouring β-lactamases encoding genes isolated from Brazilian soils. J Appl Microbiol. 2018 Aug;125(2):506-512.
doi: 10.1111/jam.13885
PMID: 29675924
Eveillard M, Kempf M, Belmonte O, Pailhoriès H, Joly-Guillou ML. Reservoirs of Acinetobacter baumannii outside the hospital and potential involvement in emerging human community-acquired infections. Int J Infect Dis. 2013 Oct;17(10):e802-5.
doi: 10.1016/j.ijid.2013.03.021
PMID: 23672981
Hernández-González IL, Castillo-Ramírez S. Antibiotic-resistant Acinetobacter baumannii is a One Health problem. Lancet Microbe. 2020 Nov;1(7):e279.
doi: 10.1016/S2666-5247(20)30167-1
PMID: 35544213
Zheng XR, Zhu JH, Zhang J, Cai P, Sun YH, Chang MX, Fang LX, Sun J, Jiang HX. A novel plasmid-borne tet(X6) variant co-existing with blaNDM-1 and blaOXA-58 in a chicken Acinetobacter baumannii isolate. J Antimicrob Chemother. 2020 Nov 1;75(11):3397-3399.
doi: 10.1093/jac/dkaa342
PMID: 32766775
Hernández-González IL, Mateo-Estrada V, Castillo-Ramirez S. The promiscuous and highly mobile resistome of Acinetobacter baumannii. Microb Genom. 2022 Jan;8(1):000762.
PMID: 35075990